Comparative genomics reveals complex natural product biosynthesis capacities and carbon metabolism across host-associated and free-living Aquimarina (Bacteroidetes, Flavobacteriaceae) species
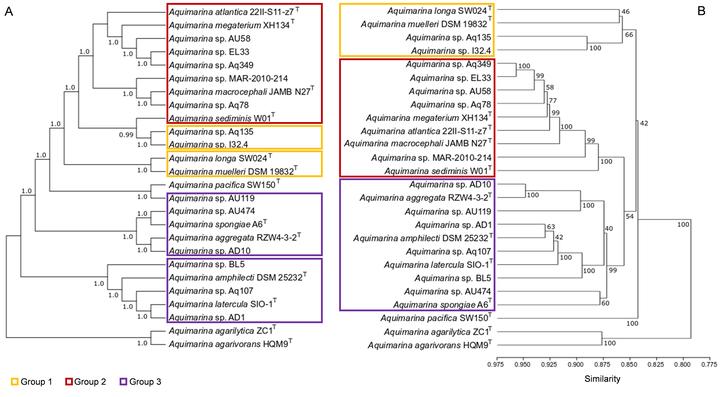 16S rRNA phylogenetic tree versus COG-based dendogram.
16S rRNA phylogenetic tree versus COG-based dendogram.Abstract
This study determines the natural product biosynthesis and full coding potential within the bacterial genus Aquimarina. Using comprehensive phylogenomics and functional genomics, we reveal that phylogeny instead of isolation source [host‐associated (HA) vs. free‐living (FL) habitats] primarily shape the inferred metabolism of Aquimarina species. These can be coherently organized into three major functional clusters, each presenting distinct natural product biosynthesis profiles suggesting that evolutionary trajectories strongly underpin their secondary metabolite repertoire and presumed bioactivities. Aquimarina spp. are highly versatile bacteria equipped to colonize HA and FL microniches, eventually displaying opportunistic behaviour, owing to their shared ability to produce multiple glycoside hydrolases from diverse families. We furthermore uncover previously underestimated, and highly complex secondary metabolism for the genus by detecting 928 biosynthetic gene clusters (BGCs) across all genomes, grouped in 439 BGC families, with polyketide synthases (PKSs), terpene synthases and non‐ribosomal peptide synthetases (NRPSs) ranking as the most frequent BGCs encoding drug‐like candidates. We demonstrate that the recently described cuniculene (trans‐AT PKS) BGC is conserved among, and specific to, the here delineated A. megaterium‐macrocephali‐atlantica phylogenomic clade. Our findings provide a timely and in‐depth perspective of an under‐explored yet emerging keystone taxon in the cycling of organic matter and secondary metabolite production in marine ecosystems.
Sandra Godinho Silva
PhD candidate
Training bioinformatician with a special interest in (meta)genomics.